Tras ver lo bien que funcionaba VQGAN+CLIP para generar imágenes yo también quería hacer un programa que creara arte a partir de un texto pasado como referencia. Por supuesto el resultado ha sido un fracaso, sin embargo se puede aprender de los fracasos.
La idea
La idea es usar solo CLIP con algoritmos genéticos. CLIP permite calcular (más o menos) la diferencia entre una imagen y un texto. Si le paso el texto «pato» y una foto me dice como de parecido es el contenido de esa foto al texto, en este caso a un pato. La idea es usar CLIP como función fitness. Para ello calculamos el vector que CLIP asocia a la imagen generada por nuestro algoritmo genético (luego vemos esto) y el vector que asocia a nuestro texto de referencia y calculamos la similitud coseno . La similitud coseno devuelve un valor entre -1 y 1:
1 significa que es idéntico
0 significa que no tiene nada que ver (es ortogonal)
-1 que es opuesto
Por lo tanto nuestra función fitness seria:
fitness = 1 - simCos(vectorTexto, vectorImagen)
Vamos a ver un poco de código.
Vector de características de texto:
#vector de carateristicas de texto
text_tokenize = clip.tokenize(TEXT).to(device)
with torch.no_grad():
text_features = model.encode_text(text_tokenize)
text_features /= text_features.norm(dim=-1, keepdim=True)
Vector de características de la imagen:
#vector de carateristicas de la imagen
image = preprocess(im).unsqueeze(0).to(device)
with torch.no_grad():
image_features = model.encode_image(image)
image_features /= image_features.norm(dim=-1, keepdim=True)
Teniendo una función fitness ya puedo usar cualquier metaheurística. En este caso se trata de optimizar una imagen para que se acerque cada vez más a lo que CLIP considera similar al texto.
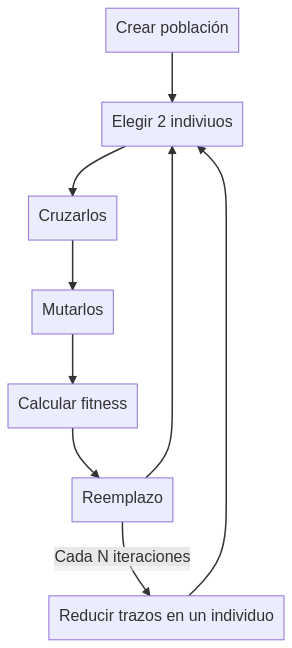
Para este caso vamos a usar algoritmos genéticos. Cada individuo es una imagen. Por lo tanto la población es un conjunto de imágenes que irán evolucionando y cruzándose compitiendo por reducir el valor de la función fitness.
Individuos, cruces y mutaciones
En un primer momento intente que el genotipo fueran los pixeles de la imagen y que las mutaciones y cruces fueran cambios aleatorios en los mismos. Manejar 16 millones de colores por cada pixel me parecían un espacio de soluciones muy amplio y decidí hacerlo en escala de grises por lo que solo tendría 256 posibilidades por pixel. Sin embargo el resultado fue un desastre los pixeles sueltos eran tan «pequeños» que creaba un «ruido» que no era capaz de evolucionar. Como solución intenté crear «píxels» más gordos. Para ello use cuadrados. Tampoco funcionó. Añadir colores dio como resultado cosas que podían tener sentido pro dentro de un ruido de colorines.
Siguiente plan, separar el genotipo y el fenotipo. El genotipo serían las instrucciones para construir la imagen mientras que el fenotipo sería la propia imagen. Como pasos para construir la imagen decidí usar pinceladas. En este caso para simularlas use líneas rectas que van de un punto aleatorio a otro, con color y anchura también aleatorios.
Una vez tenemos definidos los individuos hay que definir sus operaciones de cruce, de mutación y de reemplazo.
Operadores de cruce:
Intercambiar un bloque de trazos
Mezclar todos los trazos
No cruzar
Operadores de mutación:
Mover un trazo
Cambiar de color un trazo
Cambiar de orden un trazo por otro
Añadir un trazo
Como operador de reemplazo se reemplaza al peor individuo de la población (fitness más alto) por aquel hijo que tenga menos fitness que él.
Como el resultado era caótico, lleno de lineas de colores que no aportaban nada a CLIP pero dificultaba que un humano reconociera el dibujo decidí minimizar el número de trazos, para ello cada cierto tiempo se ejecuta una función de optimización local que prueba a eliminar cada uno de los trazos y si no altera o reduce la función fitness lo descarta.
Como último añadido y para dar más variedad a los trazos incluí que un trazo pueda ser una linea, un rectángulo o una elipse. Y una función mutación que cambia el tipo de trazo.
El resultado es un artista minimista que trata de trazar el menor número de líneas para dibujar un concepto.
Una Inteligencia artificial conceptual y minimalista
Hemos logrado un artista que crea con el menor número de trazos posible. Conceptual y minimalista…solo hay un problema que es un autor conceptual y minimalista desde el punto de vista de una IA. Hay imágenes en la que coincidimos, por ejemplo con las rosas:
Rosa
Otras imágenes sin embargo son incomprensibles, por ejemplo cuando le pido que me dibuje un pato le basta con dibujar una linea naranja rodeada de lineas verdosas, me lo hizo varias veces.
Pato ¿?
¿Por qué esa linea es un pato? Seguramente porque lo identifica con el pico y no necesita más para identificar un pato. Así que como nuestro autor minimalista reconoce patos por el pico (su rasgo más distintivo) es difícil que surja una mutación que mejore ese resultado.
Algo parecido me pasa con los dragones que no me pinta el cuerpo solo la cabeza, generalmente con algún tipo de «cresta» y a veces las patas, pero nada de cuerpo. Lo cual tiene sentido ya que su cuerpo se parece al de otros reptiles.
¿El dragón sin cuerpo?
Como herramienta puede ayudarnos a entender que «ve» una IA en una imagen.
Para probarlo dejo este enlace al codebook de Google donde probarlo
Esto es una introducción a las bases biológicas que inspiran muchos algoritmos. Debo advertir que es una introducción escrita por un informático para gente interesada en algoritmos así que los que sepáis de biología no seáis muy duros. Soy consciente de que he hecho muchas simplificaciones.
Que algunos algoritmos estén inspirados no significa que sean copias exactas, no siempre la solución aportada por la naturaleza es la más sencilla de llevar acabo, o la que mejor se adapta a nuestros problemas por eso lo que se busca es imitar los métodos para conseguir los mismos resultados que la naturaleza. Una vez conseguida la imitación esta se puede modificar, variar o combinar para dar origen a métodos jamas intentados por la naturaleza.
Un único modelo
Actualmente el único modelo que tenemos de seres vivos es el de aquellos que nos rodean. Esto nos «limita», ya que pese a su gran variedad toda la vida proviene de un origen en común. Nuestra definición de vida y sus características se han de basar en un único modelo.
La base de este modelo es la célula, esta es la forma de vida más simple (los virus no se considera que esten vivos), todos los seres vivos están compuestos por ellas, ya sean unicelulares o pluricelulares. Es por así decirlo el ladrillo con el que se construye la vida.
La célula
Estudiar la célula en toda su complejidad daría para varios textos como este. Para que nos sea más fácil olvidaremos el resto de funciones de la célula y la veremos como una maquina destinada únicamente a trabajar con la información del ADN.
La célula esta formada por una membrana que la separa del medio, en su interior se encuentra el citoplasma, que se divide en hialoplasma y organulos citoplasmáticos. El hialoplasma es medio que contiene a los organulos citoplasmaticos, en el se realizan numerosas reacciones químicas, esta formado en un 70% de agua y el resto son proteínas, lípidos, glucidos, etc. Los organulos citoplasmáticos desempeñan diversas funciones dentro la célula. Nosotros vamos a centrarnos en el núcleo.
El núcleo y los cromosomas
En el interior del citoplasma, separado por la membrana nuclear, se encuentra el núcleo de la célula con toda la información genética. No todas las células tienen un núcleo, en algunos casos como las algas verdeazuladas, y las bacterias, el material genético se encuentra libre en el citoplasma, las células se dividen en dos grupos según posean o no núcleo diferenciado:
Procariotas: Carecen de núcleo diferenciado el material genético se encuentra en el citoplasma.
Eucariotas: Poseen una estructura separa del citoplasma donde están los cromosomas, a esta estructura se le denomina núcleo.
El núcleo esta formado principalmente por proteínas denominadas histones y ADN que forman los cromosomas. Estos solo son visibles durante el proceso de división celular cuando se enrrollan para formar la conocida estructura con forma de X
En la mayoría de las células de los animales y plantas poseen células diploides, es decir, con dos copias de cada cromosoma. Las células con un solo juego de cromosomas se conocen como haploides. En la reproducción sexual cada progenitor aporta un cromosoma de cada par. Sabiendo esto es fácil imaginarse que las células reproductoras (gametos) serán haploides, ya que el único cromosoma por par que poseen se juntara con el del otro gameto para formar una célula diploide. Cada especie tiene un numero distinto de cromosomas.
ADN y ARN:
El ADN es una molécula constituida por dos largas secuencias de nucleótidos, ambas se enrollan sobre si mismas formando una espiral, están unidas por puentes de hidrogeno. La información esta codificada por cuatro nucleótidos; adenina (A), timina (T), guanina (G) y citosina (C) en el caso del ADN, en el ARN se sustituye la timina por el uracilo (U). Los nucleótidos se unen unos frente a otros para unir ambas cadenas de nucleotidos, la adenina se una con la timina y la citosina con la guanina de tal modo que si tenemos la secuencia ATGCA sé unirá con la TACGT. Esto significa que ambas cadenas son complementarias y que se puede codificar una a partir de la otra.
Los grupos de tres nucleotidos, se denomina codón, sirven para identificar los distintos aminoácidos, piezas estructurales de lo polipéptidos que a su vez se unen para formar proteínas. No se usan mas de veinte aminoácidos distintos, por eso hace falta tres nucleótidos par definir un aminoácido ya que si fueran dos solo habría 16 (42) combinaciones distintas, con tres se dispone de 42 (43) combinaciones. Además algunos codones se usan para guiar el proceso de transcripción del ADN, indicando donde acaba el gen (STOP) o donde empieza (START). Como sobran combinaciones varios aminoácidos están definidos por más de un codón.
1ª
2ª
2ª
2ª
2ª
3ª
T
C
A
G
T
Fenilanina
Serina
Tirosina
Cisteína
T
T
Fenilanina
Serina
Tirosina
Cisteína
C
T
Leucina
Serina
STOP
Selenocisteina STOP
A
T
Leucina
Serina
Pirrolisina STOP
Triptófano
G
C
Leucina
Prolina
Histidina
Arginina
T
C
Leucina
Prolina
Histidina
Arginina
C
C
Leucina
Prolina
Glutamina
Arginina
A
C
Leucina
Prolina
Glutamina
Arginina
G
A
Isoleucina
Treonina
Asparagina
Serina
T
A
Isoleucina
Treonina
Asparagina
Serina
C
A
Isoleucina
Treonina
Lisina
Arginina
A
A
Metionina START
Treonina
Lisina
Arginina
G
G
Valina
Alanina
Ac.
Aspártico
Glicina
T
G
Valina
Alanina
Ac.
Aspártico
Glicina
C
G
Valina
Alanina
Ac.
Glutámico
Glicina
A
G
Valina
Alanina
Ac.
Glutámico
Glicina
G
El ADN codifica la información siguiendo un patrón: el inicio de la información útil es indicado por el codón ATG, a partir de ese momento si este codón vuelve a salir será interpretado como el aminoácido metionina, la traducción de codón a aminoácido sigue hasta llegar a un codón de STOP representados por TAA, TAG, TGA, aunque hay seres vivos donde los dos últimos también sintetizan aminoácidos.
Para que el ADN se traduzca de información a proteínas o a enzimas hace falta que este sea leído y transcrito; vamos a ver como se lleva a cabo este proceso. Se inicia con la transcripción de ADN a ARNm (ARN mensajero). La cadena de ADN se abre y permite que una enzima, la ARN-polimerasa copia el fragmento de ADN traduciendolo a ARNm. El fragmento es complementario con la timina sustituida por el uracilo (recuerda que es ARN) de manera que la cadena AGTCG se codificará como UCAGC.
El ARNm forma una larga y delgada cadena, esta se inserta en los ribosomas que actuan como pequeños automatas recorriendo la cadena de ARNm leyendola y ensamblando aminoacidos hasta llegar al final del gen indicado por un simbolo de STOP (UAA, UAG, UGA). La cadena de ARNm no es recorrida por un solo ribosoma sino que son varios al mismo tiempo. En el ribosoma se encuentra ARNt (ARN de transferencia), que detecta los codones usando la forma complementaria (anticodón), así el ribosoma sabe que aminoácido unir a la cadena. En el caso de las células procariotas, los ribosomas pueden empezar a trabajar antes de que se acabe de traducir el ADN a ARNm. La secuencia de nucleotidos comprendidos entre un START y un STOP definen una enzima o una proteína especifica y se denomina gen o locus.
Genes
Los genes son la unidad mínima de información hereditaria, codifican una proteína o una enzima. Hay que distinguir dos conceptos; el genotipo, la secuencia de ADN de un individuo, y el fenotipo, la manifestación física del genotipo. En individuos con un solo gen el genotipo corresponde con el fenotipo, pero en el caso de los seres con células diploides se plantea el problema de elegir cual de los dos genes se va a usar como modelo. Para solucionar esto los genes poseen distintos “pesos”. Cuando un gen se impone sobre otro se dice que es dominante y se representa con una letra mayúscula, en caso de que el gen sea enmascarado por el otro se dice que es recesivo y se representa con una letra minúscula. A cada una de las variaciones que presenta un gen se le denomina alelo.
Veamos un ejemplo, el albinismo esta relacionado con un gen en particular, es recesivo y el alelo se representa por a, la pigmentacion normal de la piel esta relacionada con el alelo A y es dominante. Por los que hay cuatro combinaciones, que realmente son tres ya que una esta repetida.
AA – Pigmentación normal, ambos genes son dominantes.
Aa – Pigmentación normal, no sufre albinismo pero es portador de la enfermedad
aA – Pigmentación normal, no sufre albinismo pero es portador de la enfermedad
aa – Albino, al tener ambos cromosomas recesivos el albinismo se manifiesta ya que no hay ningún gen para enmascararlo.
Se puedes dar el caso de que lo genes sean codominantes, ninguno domina sobre el otro, y ambos se manifiestan. Un ejemplo lo tenemos en el dondiego de noche una planta que tiene dos alelos para el color de sus flores: rojo y blanco (genotipo). Sin embargo muestra flores de tres colores: blancas, rojas y rosas (fenotipo). Las rojas y las blancas tienen ambos genes del mismo alelo (rojo o blanco), mientras que las flores rosas se produces por la codominancia de ambos alelos (rojo y blanco)
Este no es el único modelo, muchas veces un carácter esta determinado por varios genes, o un gen determina más de un carácter, un ejemplo es la herencia cuantitativa, la idea es que por ejemplo cinco genes determinaran el tamaño de una planta, un gen recesivo r hace que crezca 5 centímetros y uno dominante R que crezca 10 centímetros, así que una planta rR rr RR medirá (10+5+10) 25 centímetros y otra Rr rr rr (10+5+5) 20 centímetros se pueden ver que plantas de distinto genotipo tendrán igual fenotipo, por ejemplo rr RR Rr y RR RR rr. En la vida real los caracteres no están determinado de una manera tan sencilla y lineal.
Los genes no solo cumplen una simple función de almacén de información, también tienen una función reguladora, hay grupos de genes que se ocupan de bloquear o desbloquear la producción de enzimas y proteínas dependiendo de la cantidad de estas. Para ello un gen conocido como regulador produce una proteína represora que funciona de la siguiente manera. En cada gen existe un segmento de ADN llamado promotor al cual se adhiere la ARN-polimerasa, entre este segmento y el gen a veces existe otro segmento de ADN llamado operador al que se liga la proteína represora y evita que la ARN-polimerasa puede generar el ARNm. Dependiendo de la concentración en el medio de ciertas sustancias la proteína represora puede ser inhibida por ellas y no llegar a unirse al operador. Este mecanismo se haya muy estudiado en las bacterias, los seres vivos superiores poseen un mecanismo más complicado y todavía no muy bien comprendido en el que intervienen proteínas ligadas a proteínas que estimulan la transcripción de ADN, en esencia el resultado conseguido es el mismo. A los genes que forman el sistema de regulación completo se les denomina en conjunto operón.
A su vez a otros genes con distintas funciones, como los genes de segregación cuya función es estructural y sirven como punto de apoyo para la construcción de los cromosomas y en la división celular. Y genes sin función aparente conocidos como ADN basura, que es codificado por la ARN-polimerasa en ARNm pero que es desechado por los ribosomas durante la fase de transcripción. El ADN se divide en dos tipos; exones, que es el ADN portador de información y que se codifica en ARNm e intrones, formado por secuencias de ADN no codificante que se intercalan entre los exones dentro de los genes. Actualmente no se conocen todavía todas las funciones de los intrones, presentes solo en las células diploides, excepto los gametos que son haploides. La mayor parte de estos están formados por repeticiones de nucleotidos o por zonas con gran facilidad para recombinarse o como los transposones para saltar de gen en gen o incluso de cromosoma en cromosoma, lo que puede inducir mutaciones.
Un descubrimiento aun no muy bien entendido, es que un mismo gen puede codificar distintas proteínas, el truco esta en que no todos los exones de un gen se codifican para forma una proteína, sino que lo hacen distintas combinaciones de estos, de tal forma que un gen puede llegar a codificar varias de proteínas.
La replicación de los genes
El ADN posee la capacidad de autorreplicarse, puede sacar copias de si mismo y, por lo tanto, de toda la información que porta. Como hemos visto el ADN se compone de dos copias de cada gen. La duplicación de una hebra de ADN da lugar a otra, pero no siempre son iguales, los genes se pueden mezclar entre ellos y cambiar de hebra, dando lugar a hebras con genes que sean mezcla de ambos progenitores.
No es necesario entrar en detalles de cómo se replica el ADN para obtener una copia exacta (ya veremos que no siempre es así) de los genes. Durante la copia se da un proceso conocido con el nombre de sobrecruzamiento. El sobrecruzamiento consiste en que dentro de una pareja de cromosomas los genes de ambos progenitores se mezclan para dar lugar a una cadena de ADN que contiene genes de ambos. Este cruce solo se da dentro de una pareja de cromosomas, y los cruces se hacen, según parece, al azar. El mecanismo es el siguiente, un grupo de genes es separado y ligado con otro grupo para formar una sola cadena de genes, como podréis imaginar a mayor proximidad física de los genes más fácil es que sean heredados juntos,. A los genes que son heredados juntos se dicen que están ligados. Los genes ligados solo se pueden dar en caso de que todos pertenezcan al mismo cromosoma. Hay que tener en cuenta, que dos genes no pueden estar ligados sin que también lo estén los genes que haya entre ellos.
De esta manera a partir de dos cadenas diferentes, sometiéndolas a sobrecruazamiento se obtienen una gran cantidad de cadenas distintas, tan solo combinando genes. Este número es verdaderamente grande, si pensamos que para cada gen hay 2 posibilidades, lo que quiere decir que para un gen habrá 2 combinaciones, para 2 genes habrá 4 (2*2) combinaciones, para 3 genes habrá 8 (2*2*2) combinaciones, para 4 genes habrá 16 (2*2*2*2) combinaciones. Es decir para un numero n de genes habrá 2n combinaciones distintas.
Pero si no hay ninguna variación en los genes. ¿De dónde salen los nuevos alelos?
Mutaciones:
Los genes son simples estructura químicas, que actúan como un mapa para montar aminoácidos. Supongamos que un codón cambia una base por otra, CTT por CTA, en ambos casos sintetiza el aminoácido Leucina. Pero si en lugar de cambiar la tercera base cambia la primera TTT tenemos Fenilanina. Un pequeño cambio puede alterar la estructura de la proteína a sintetizar.
¿Pero como puede darse este cambio de una base por otra?. Las razones pueden ser varias, desde errores en el mecanismo de replicación del ADN hasta causas externas, como la radiación, que alteren las bases. La mayor parte de estas alteraciones, conocidas como mutaciones, no serán viables pero las pocas que si lo sean permitirán crear un individuo con un nuevo alelo para el gen.
Como la mayoría de las mutaciones son nocivas hay mecanismos para reducirlas. Mientras el ADN se replica hay enzimas que realizan una comprobación de que la copia es correcta y en caso de detectar un error lo corrigen. Su eficacia, aunque alta, no es del cien por cien. Como podemos ver el equilibrio es difícil, si se producen muchas mutaciones aparecerán nuevos alelos, pero si se producen demasiadas alguna será letal y las demás mutaciones se perderán ya que el individuo no prosperara.
Evolución
Cuando una mutación resulta en un individuo válido está mutación puede resultar beneficiosa, perjudicial o neutral.
Los seres vivos desarrollan su actividad dentro de un ecosistema, su interacción con este es lo que decide si una mutación le da o no una ventaja al individuo, vamos a ver un ejemplo. La anemia falciforme es una enfermedad de origen genético, causada por el cambio de una única base en un gen. Si ambos genes tienen esa mutación los glóbulos rojos adopten forma de media luna. Estos globulos rojos son menos flexibles y tienen una vida útil menor que los normales lo que causa anemia. En un principio parece ser una mutación que da al individuo una clara desventaja, excepto en algunos lugares de África donde además de la anemia hay otra amenaza mayor el paludismo. La gente con anemia es mucho mas resistente al paludismo, por lo que en muchos pueblos afectados por el paludismo se encuentra gran cantidad de gente que sufre de anemia falciforme y donde una mutación que parecia una desventaja se ha convertido en ventajosa.
Podemos ver que los individuos pueden poseer ventajas que les permita vivir más tiempo o mantenerse en mejor estado, esto les facilita tener más descendencia, esta descendencia será portadora de sus genes y por lo tanto podrá poseer también esa ventaja, lo que a su vez les permitirá vivir más y tener más hijo. Esto hará que ese alelo se extienda por toda la especie. Esto es una pequeña parte de la teoría de la evolución de Darwin-Wallace. Todo esto se podría resumir con la expresión: “La supervivencia del más apto”.
La nueva mutación puede ser dominante o recesiva, dando lugar a distintos casos. Primero supongamos que es dominante y produce una desventaja, todos los individuos con esa mutación lo tendrán más difícil para tener descendencia y es probable que ese alelo desaparezca. En el segundo caso vamos a suponer que es dominante y produce una ventaja, ese alelo se extenderá a gran velocidad. Para el tercer caso vamos a suponer la mutación recesiva y ventajosa, tardara más en extenderse, pero los otros alelos dominantes al representar una desventaja iran desapareciendo. El ultimo caso es el más interesante, la mutación es recesiva y desventajosa, parece que tiene todas las de perder, pero no es así, al «quedar oculta» por otro alelo dominante y ventajoso esta mutación puede permanecer “escondida”. Solo reaparecerá en casos en que se encuentren dos alelos iguales.
Ahora bien, el medio puede cambiar y convertir a individuos aptos en individuos poco aptos, en ese momento los alelos más ventajosos se pueden convertir en una carga para su portador y los alelos desventajosos convertirse en una ventaja. En esas circunstancias los alelos recesivos “escondidos”, pueden volver a aparecer, solo que convertidos en ventaja y extenderse rápidamente. Un caso así lo tenemos en Londres, al principio de la época industrial, en las afueras había un grupo de polillas que podían ser de dos colores, blanco o negro, en un principio las polillas blancas tenían ventaja pues vivían en un bosque de abedules blancos y lo tenían fácil para confundirse con sus troncos y evitar a los depredadores y su color era el mas abundante, pero con la llegada de las fabricas y sus hornos de carbón los arboles se tiñeron de negro y la población cambio, rápidamente el número de polillas negras supero al de blancas. Vemos el medio afecta al rumbo de la evolución.
Podemos pues decir que la evolución no tiene un rumbo marcado, simplemente se adapta al medio, por lo que los términos superior inferior carecen de sentido, y habría que cambiarlos por mejor o peor adaptado. Esta adaptación provoca un curioso efecto, si dos poblaciones idénticas son separadas y colocadas en entornos distintos cada uno evolucionara por caminos diferentes, es lo que se conoce como deriva genética.
Para que una especie pueda adaptarse ha de tener variedad, sus individuos han de presentar múltiples características distintas, cuantas más, mejor, eso permite que se puedan presentar gran cantidad de soluciones adaptativas a un cambio en el entorno. Para una especie con poca variación genética cualquier cambio brusco del medio puede suponer su extinción. Si en el caso de antes de las polillas todas hubieran sido blancas seguramente habrían desaparecido.
Actualmente aun se duda de si la evolución produce avances significativos de golpe, “saltos evolutivos”, o si realiza su trabajo lentamente. Lamentablemente el registro fósil parece apoyar ambas teorías. Aunque cada vez cobran mas fuerza las teorías sobre sistemas caóticos en equilibrio que van acumulando pequeñas mutaciones hasta que este cumulo de mutaciones produce un gran cambio.
Es interesante mencionar una teoría anterior a la actual teoría de la evolución, la teoría Lamarckiana, según la cual a lo largo de la vida un animal desarrolla alguna característica concreta debido a su uso, esta característica pasa a sus descendientes que también la desarrollan y a su vez les da mayor ventaja frente a los otros individuos. Parece fuera de lugar describir esta teoría aquí, puesto que ha sido demostrada errónea y no se corresponde con todo lo explicado hasta ahora del ADN, pero hay algoritmos que sacan partido de ella sometiendo a los individuos a un proceso «de mejora» entre generaciones.
Modelo biológico de lo algoritmos genéticos
Los algoritmos genéticos se pueden implementar de gran variedad de formas, pero generalmente se sigue un mismo modelo que simplifica mucho la implementación, aquí se va a intentar caracterizar a ese modelo con las distintas estrategias de los seres vivos.
Se suele usar una sola cadena, por los que se podría decir que es haploide, a su vez los individuos son asexuados ya que todos se pueden cruzar entre si. La selección se basa en el fenotipo que se obtiene a partir del genotipo, por lo que se acerca al modelo de Darwin-Wallace. El tamaño de la población es fijo y estable, los individuos menos aptos son sustituidos por otros más aptos, por lo que existe competencia. Los individuos están sometidos a mutaciones aleatorias.
Teniendo en cuenta estas características quizás el modelo biológico al que mas se parecen los algoritmos genéticos sea el de una cepa de bacterias, ya que poseen una única hebra de ADN, poseen una alta tasa de mutación y de adaptabilidad, son capaces de intercambiar fragmentos de ADN. Dentro de la cepa las más aptas se extienden rápidamente y van sustituyendo a las menos aptas.
Otras vías de herencia
Hasta aquí es lo que se suele contar como base para los algoritmos genéticos y evolutivos, pero no hay que olvidar otras formas de herencia que también pueden tener su utilidad.
La primera serian las mitocondrias, pequeños orgánulos que actúan como centrales energéticas de las células y están dotadas a su vez de ADN, conocido como ADNm, estos orgánulos se heredan de la madre en la mayoría de las especies, lo que parece contradecir todo lo dicho hasta ahora, pero así es, una parte de nuestra herencia proviene únicamente de la madre, luego sus ventajas o desventajas provienen únicamente de un progenitor sin posibilidad que el otro intervenga. De hecho la evolución del ADNm se puede estudiar separada de la del ADN del resto de la célula.
El segundo caso lo representan las bacterias, que actúan como redes de intercambio de genes, los genes son capaces de separarse y recombinarse con los de otras bacterias. Se ha estudiado este fenómeno desde el punto de vista de la vida artificial usando autómatas celulares.
La tercera vía son los virus, a grandes rasgo, algunos son capaces de insertar en el ADN de la célula fragmentos del suyo para reproducirse, lo que a su vez puede producir el proceso contrario y que parte del ADN de la célula huesped pase a los nuevos virus. Actualmente se usan como vectores para terapia genética.
La cuarta vía son las proteínas, capaces de transformar otras proteínas en copias de si mismas, que a su vez transformaran a otras en lo que se denomina una reacción autocatalizada. Este tipo de proteínas se denominan priones y son la causa de enfermedades como la de las «vacas locas».
Una quinta sería la clonación o replicación exacta del individuo, es muy usada entre los insectos ya sea para formar colonias o como alternativa a la reproducción sexual en forma de partenogénesis cuando hay escasez de machos. En esta forma la única fuente de variación genética son las mutaciones.
Por ultimo repetir que lo aquí visto no es mas que una introducción y que los métodos de codificación del ADN aun no han sido del todo desentrañados, pero ya se han empezado a encontrar sistemas bastante complejos como cadenas que ADN que codifican en ambos sentidos, cadenas de ARNm que son “asaltadas” y modificadas antes de llegar a los ribosomas, genes que se solapan, microARN (mARN) e interruptores ribosomaticos. No esta de más recordar que la complejidad de una solo célula supera en muchos niveles a la complejidad de cualquier construcción creada por el hombre. Pero toda esta complejidad no hay que verla como un obstáculo ya que cada nuevo proceso o parte que se desentraña, no hace otra cosa que dar más material sobre el que trabajar, más ideas y opciones.
Si ajustar una metaheurísticas ya cuesta. ¿Por qué querríamos sufrir el doble para hacerlo con dos?. Hay varios motivos, el principal es que una metaheurística no se comportan bien en todo el espacio de soluciones del problema y por ello se usan varias.
Un caso es que una metaheurística se puede comportar muy bien para calcular parte de la solución pero no toda. Entonces algunos de sus valores los calcularemos usando una metaheurística dejando intactos aquellas partes de la solución para los que no funciona a los que posteriormente aplicaremos la segunda metaheurística que al igual que la anterior no alterará la parte para la que no funciona. Este proceso tiene que repetirse varias veces permitiendo que ambas partes evolucionen de forma conjunta.
Otro caso en que resulta útil aplicar una segunda metaheurística es cuando tenemos una que funciona muy bien explorando el espacio de soluciones y encontrando la zona donde se encuentra el máximo pero muy mal para precisar el valor de este. En ese caso se usa una metaheurística para acercarnos al máximo y cuando ha terminado aplicamos otra para precisar el resultado.
Ahora el problema es como combinar varias metaheurísticas ya que algunas funcionan con mutiles soluciones y otras con una sola.
Empezamos por el caso más intuitivo, el de que ambas metaheurísticas recurran a una población de varios individuos. Aquí las opciones son varias:
Cada metaheurística se ejecuta de forma separa con sus propia población de soluciones y cada cierto tiempo se intercambian soluciones entre ellas.
Ambas metaheurística actúan sobre las mismas soluciones y compiten entre ellas. Solo las mejores soluciones pasan a formar parte de la población
Intercalar metaheurísticas. Aplicamos una durante varias iteracciones una y luego iteramos varias veces con la otra.
Para el caso de combinar dos metaheurísticas que trabajan con un solo resultado todas las opciones se limitan a usar primero una y luego la otra. Este proceso puede repetirse varias veces o realizarse solo una vez.
Para el caso de combinar metaheurísticas con población y las que trabajan mejorando una única solución:
Tras cada ciclo de la metaheurística orientada a población se aplica la otra metaheurística a los individuos nuevos tratando de mejorarlos
Otra opción es usar la metaheristica individual como función de modificación/creación de nuevo individuos.
Un ejemplo muy habitual de combinar metahurísticas es en el caso de los algoritmos genéticos, tras haber generado los nuevos individuos usando los operadores de mutación y/o cruce y antes de que compitan con el resto de la población se trata de mejorar los nuevos individuos usando alguna metaheurísticas que explore el espacio cercano a cada individuo tratando de encontrar el máximo local.
La derivada df de una función f es otra función que expresa la tasa de cambio de f. Por ejemplo si tenemos una función que calcula la velocidad en el punto X y calculamos su derivada tendremos una función que calcula la aceleración (cambio de la velocidad) en el punto X. Otra forma de entenderla es como la pendiente en un punto. Si calculamos la derivada de la función fitness en un punto tendremos una función que nos dice cuánto varía y si aumenta o disminuye. Es decir nos da una pista clara de en qué sentido hay que moverse para aumentar o disminuir el valor de la función.
Esto también quiere decir que si la pendiente es 0 ya estás en un máximo o en un mínimo. Si calculas todos los puntos donde la derivada es 0 seguro que uno de ellos es el máximo o el mínimo que estás buscando. Desgraciadamente en muchos casos no es sencillo hallar esos puntos.
Siendo una herramienta tan útil vamos a ver cómo calcularlo. A muchos os sonará de haberlo estudiado y haber tenido que memorizar un montón de derivadas y luego aprender reglas de derivación (por partes, cambio de variables,…) Incluso puede que tener pesadillas con ellas. Pero los ordenadores no saben hacer ese tipo de derivadas. Con ellos hay que recurrir derivadas numéricas que son mucho más simples.
df(x) = f(x+h) – f(x-h) / 2*h
¿Cuanto es h? Lo ideal es que sea una cantidad lo más pequeña posible. Pero ojo con cantidades muy pequeñas y la precisión numérica de los lenguajes de programación que puede dar sorpresa. Cómo se puede ver en el código más abajo yo he fijado el valor por defecto a 0,001.
Para ir más allá de la primera derivada también hay fórmulas para calcularla numéricamente que son más exactas que el método que vamos a usar nosotros pero en este caso me ha parecido más interesante el siguiente método.
Tenemos una función diff(x,f) que calcula la derivada de f en el punto x. Si hacemos diff(3,diff(3,f)) calcularíamos la segunda derivada de la función f en el punto 3. Para la tercera tendríamos que hacer diff(3,diff(3,diff(3,f))). Para las siguientes derivadas creo que ya habéis entendido como funciona.
En lugar de escribir todas las funciones una detrás de otra en una larga cadena vamos a usar una función que lo haga por nosotros. Siendo el parámetro n el número de derivada que calculará.
Ya hemos visto como funciona la derivada para una función f(x) en un punto A, ahora os estaréis preguntando «¿Cómo funciona la derivada para una función con más variables como f(x1, x2)?» ¿A qué os he leído la mente?. Esta vez el punto donde se calcula tiene dos coordenadas [A,B]. Se calculan dos derivadas respecto de x1 y respecto de x2. Lo de «respecto» significa que solo se va a calcular modificando esa varíable, la otra será estática.
df(x1,x2)/dx1 = f(x1-h,x2)-f(x1+h,x2)/2*h
Es decir dejamos el resto de las «equis» inalteradas.
El gradiente es el cálculo de la derivada respecto a «cada equis». Es decir que
G = [df(x1,x2)/dx1, df(x1,x2)/dx2];
Podemos calcular el segundo, tercer,…. gradiente calculando la correspondiente derivada.
Nuestra función para calcular el gradiente usando la función anterior para calcular la derivada numérica queda así:
this.gradN = function(n, x, f){
G = [];
for(var i = 0; i < x.length; ++i){
var auxX = x.slice(0);
var fdx = function(x){auxX[i] = x; return f(auxX);}
G.push(this.diffN(n, auxX[i], fdx));
}
return G;
}
Para que el uso sea más intuitivo vamos a hacer una versión del primer gradiente sin el parámetro n (n=1)
Si ya hemos elegido nuestro algoritmo favorito y la función fitness que vamos a usar, lanzamos nuestro programa y surge un problema ¿Cuando páramos?.
Lo fácil sería decir que cuando alcancemos el resultado deseado, pero eso no es siempre posible o al menos no en un tiempo que sea razonable. Así que tendremos que quedarnos con una buena aproximación. ¿Pero cual es una buen aproximación? Hay veces que lo sabemos, sabemos que resultado nos tiene que dar al función fitness para que la solución sea suficiente para nosotros. ¿Pero que pasa si no sabemos cuando ha llegado al máximo o al mínimo de la misma?. Con las metaheurísticas siempre nos quedaremos con la duda. No podemos saber si hemos encontrado el punto optimo o nos hemos quedados atrapados en un máximo/mínimo local. Todo dependerá de nuestras necesidades y conocimiento del espacio de búsqueda.
Para reducir este problema hay que intentar explorar lo máximo posible el espacio de búsqueda. El punto optimo puede esconderse en cualquier rincón que nos dejemos sin mirar. Pero claro si estamos usando una metaheurística es porque no conocemos el espacio de búsqueda y es muy grande como para que lo exploremos.
Una de las condiciones más usadas en el caso de desconocimiento total es fijar de antemano un número de iteracciones. Es útil cuando tenemos un tiempo o recursos limitados para calcular la solución. Sin embargo puede darse el caso de que el tiempo no sea tan limitante, aunque tampoco nos apetezca tener al algoritmo iterando durante varias veces la vida del universo. En ese caso se puede fijar un umbral de «mejora». De tal manera que si durante un número determinado de iteracciones el resultado no ha mejorado la solución por encima de ese umbral se para la ejecución ya que se considera que el tiempo invertido no compensa la mejora obtenida.
Por último la que me atrevería a decir que es la condición más usada pero que ningún libro habla de ella. Ir mostrando datos en pantalla y que el usuario decida cuando terminar la ejecución. Puede parecer una solución «poco inteligente» pero durante las pruebas de un algoritmo para ver qué tal se comporta con un problema o viendo si se puede afinar su configuración puede ahorrar mucho de tiempo.
De todas formas muchas veces nos obsesionamos con encontrar el resultado optimo y en la vida real muchas veces basta con una aproximación que ni siquiera sea muy buena.
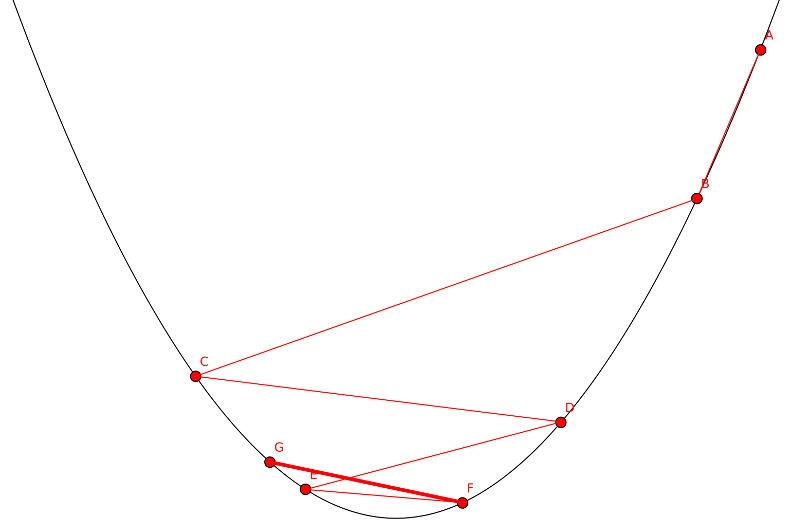
Si hay una metaheurística de ascender montañas tiene que haber otra de bajarlas. El descenso del gradiente se basa en usar el gradiente de guía para encontrar el mínimo de una función. El gradiente indica la pendiente máxima en un punto y hacia donde tenemos que dirigirnos para ascender (a favor del gradiente) o descender (en contra del gradiente). Sabiendo eso solo hay que moverse en la dirección indicada por el gradiente. ¿Cuanto? Ahí está el problema. Si el paso es muy grande podrías pasarte el mínimo y si es muy corto puedes acabar dando un gran número de pasos. Cómo lo de dar un gran número de pasos solo es un problema cuando lo haces con papel y lápiz y los ordenadores no se cansan de caminar (siendo cada paso una iteración del algoritmo) es preferible el segundo caso. Cada vez que se llega a un nuevo punto se calcula el gradiente en ese punto y se avanza en esa nueva dirección.
Supongamos que aún con nuestras precauciones de dar pasitos pequeños nos pasamos del mínimo. ¿Cómo podemos saberlo?. En la imagen de debajo se puede ver un ejemplo. Primero empezamos a «rebotar» de un lado a otro de la función, mientras el valor decrezca vamos bien. El problema surge si el valor aumenta (en la imagen el paso del punto f al g) . En ese caso hemos de volver al punto anterior, reducir el tamaño del paso y seguir probando hasta que se obtenga un valor fitness menor.
Lo ideal sería llegar a un punto donde la pendiente sea 0 (eso indica que es un máximo o un mínimo). Pero en la practica no siempre es posible y al final hemos de depender de las condiciones habituales, llegar a una solución lo suficientemente próxima a la ideal, alcanzar cierto numero de iteraciones o llegar a un tamaño de paso tan pequeño que no merezca la pena continuar.
Ya hemos visto como calcular de forma numérica la derivada y el gradiente. Ahora hemos de aplicar ese conocimiento para calcular el gradiente de la función fitness. Como el descenso del gradiente esta pensado para minimizar, hemos de elegir una función fitness que a menor valor mejor resultado.
Cada nueva coordenada seria la antigua coordenada menos el valor de la derivada de la funcion para esa coordenada multiplicado por el tamaño del paso Kstep.
cordX = cordX – (Kstep * dF/dx)
O lo que es lo mismo en código:
var grad = diff.grad(cords, this.model.calculateFitness);
for(var i = 0; i < this.parameters.dimensions; ++i){
cords[i] -= this.parameters.step*grad[i];
}
Si el nuevo valor de fitness es menor adoptamos esas coordenadas como nueva solución, si es mayor reducimos el tamaño del paso y volvemos a probar:
La función fitness suele ser una de las partes que menos trata se habla de metaheurística. Sin embargo es de las partes más importantes, si no directamente la más. Una mala heurística puede darte una mala aproximación. Pero una mala función fitness puede darte una solución completamente acertada para un problema que no es el que tú quieres resolver.
Es mportante tener en cuenta que las metaheurísticas no resuelven el problema que tu quieres, sino el que la función fitness describe. Suena como algo obvio pero causa más quebraderos de cabeza de lo que parece. Para empezar hay veces en las que no es sencillo valorar las soluciones. Por ejemplo con problemas de combinatoria, sobre todo si hay restricciones entre los elementos, puede ser complicado decir si una solución parcial es mejor que otra y más difícil aún estimar cuánto mejor. Valorar incorrectamente estas soluciones puede causar que el algoritmo «se desvíe de su camino» o incluso que haya soluciones incorrectas con valores mejores que las correcta.
Otro problema surge cuando hay que valorar soluciones que no son válidas, por ejemplo soluciones que matemáticamente tienen sentido pero son irrealizables en la práctica. En principio lo lógico puede parecer considerarlas inválidas y no continuar por ese camino, sin embargo puede ser necesario para llegar a explorar otra zona donde si son válidas.
Rendimiento
También influye en el rendimiento del algoritmo ya que el cálculo del fitness suele ser la parte más costosa de cada iteración. Cualquier optimización de la misma repercute beneficiosamente en el tiempo que cuesta realizar cada iteración.
Precisamente por eso cabe plantearse el empezar con una versión simplificada sin las restricciones más costosas o con menos resolución y según avance el algoritmo pasar a otra más precisa y completa. Como siempre estas optimizaciones no deben afectar al «relieve» del espacio de búsqueda. En las metaheurísticas con una fase de búsqueda global y otra local también puede ser interesante usar funciones menos precisas en la búsqueda global para ahorrar tiempo.
Minimizar, maximizar, optimizar
Algunos algoritmos funcionan buscando el máximo, otros el mínimo. El resultado de la función de fitness debe de adaptarse a ello. En el caso de que tengamos una función de fitness pensada para un tipo y queramos transformarla en el otro, la forma más simple es calcular el valor máximo que puede tomar la función fitness en el espacio de búsqueda (si no es posible conocerlo exactamente habrá que estimarlo) y restarle el valor de la función fitness en ese punto. Es decir a partir de una función de fitness F la nueva función fitness será:
f'(x) = fmax – f(x)
En resumen el resultado de la función fitness aporta la información a la metaheurística para funcionar. Si la función fitness no «dibuja un mapa» adecuado para la búsqueda da igual que metaheurística se use, no va a funcionar correctamente.
Gran parte de las metaheurísticas y heurísticas tienen el paso «Elegir un nuevo vecino/solución» pero no entran mucho en detalle de cómo elegirlo. La papeleta generalmente se resuelve eligiendo el vecino con el mejor fitness o uno aleatorio. Pero no son las únicas estrategias y dependiendo el tipo de espacio de búsqueda tampoco son las mejores. Vamos a ver algunas alternativas.
¿Qué es un vecino? La respuesta depende del espacio de búsqueda y el tipo de problema y la función usada para «generar» a los vecinos. La forma más generica de definirlo que se me ocurre seria: «Son vecinos de una solución S todas aquellas soluciones que se pueden alcanzar desde S». En algunos casos queda claro quienes son los vecinos, por ejemplo en un grafo. En otros la definición de que es un vecino se va a tener que ajustar como un parámetro más del algoritmo. Por ejemplo cuando definimos el vecino de una solución como todas aquellas soluciones que estén a una distancia igual o menor que d. En este caso d se convierte en un parámetro más a tener en cuenta.
Hay que distinguir entre dos tipos de vecindarios:
Poco poblados: cuando el número de vecinos es limitado y podemos calcular el fitness de todos. Suelen ser los vecindarios asociados a problemas combinacionales o a valores discretos. Eso no quiere decir que este tipo de problemas no puedan crecer tanto que se conviertan en el siguiente tipo de vecindario. Ojo al detalle de poder calcular el fitness, si la función que lo calcula es muy costosa puede resultar más rentable tratarlos como si fueran vecindarios muy poblados.
Muy poblados: cuando el número de vecinos es infinito o tan grande que es imposible (o su coste en tiempo es demasiado alto) recorrerlos todos. Generalmente corresponde a espacios de soluciones continuos.
Antes de empezar con estrategias complicadas empecemos por la y más simple y menos costosa que además funciona en ambos tipos de vecindarios. Elegir un vecino cualquiera al azar. El problema es que puede retrasar mucho la convergencia a un óptimo.
Las estrategias son distintas dependiendo del tipo de vecindario. Vamos a empezar por los vecindarios poco poblados. En este caso las estrategias más simple que se nos puede ocurrir es la de escoger el mejor: de todas las opciones buscamos la que tiene mejor fitness y la elegimos. El problema de asta estrategia es que dificulta la exploración. Pero por otro lado reduce el tiempo que cuesta llegar al óptimo, aunque aumenta el riesgo que sea un óptimo local. Un pequeño cambio puede solucionarlo. Elegimos una al azar pero no todas tienen la misma probabilidad sino que la probabilidad de seleccionar una solución es proporcional a su fitness.
Para los vecindarios muy poblados podemos hacer un truco para seleccionar los vecinos como si fueran vecindarios poco poblados, elegimos n vecinos al azar y luego aplicamos la selección sobre esos n vecinos como si fueran los únicos del vecindario.
En el caso de espacios de soluciones continuas se suelen elegir como vecinos cualquier solución que esté a una distancia D o menor. Pero esta es la versión más simple, en lugar de elegir todos los vecinos a una distancia D con igual probabilidad podemos hacer que sea más probable seleccionar los que más cerca están o los más lejanos o los que estén a una distancia intermedia. Incluso fijar una distancia mínima. Jugando con estos valores podemos alterar el comportamiento del algoritmo (exploración/convergencia) sin modificar sus valores.
Otra forma de mejorar la selección de vecinos puede ser priorizar la dirección en la que antes ya has encontrado una buena solución esperando que sigamos mejorando en esa dirección. Por ejemplo supongamos que hemos ido del punto (5, 5) al (6, 8). Eso significa que hemos sumado el vector (1, 3). Lo primero que podríamos hacer es mirar el punto (7, 11) = (6, 8)+(1, 3). Es decir repetir el mismo paso y a partir de ahí mirar cerca de ese punto. El problema de hacerlo así es que acabamos repitiendo pasos iguales cuando realmente lo que nos interesa es solo la dirección. ¿Cómo extraerla?. Dividimos cada componente del vector por la distancia recorrida. El vector que hemos sumado ha sido (1,3). La distancia es la raíz cuadrada de la suma del cuadrado de cada elemento. SQRT(1^2+3^2) = 3.1622… Así que la dirección sería (1/3.1622, 3/1622) = (0.31, 0.93). Ya lo tenemos. Ahora podemos avanzar la distancia que queramos en esa dirección. Solo hemos de multiplicar la distancia por cada uno de los valores del vector dirección. Podemos avanzar en esta dirección hasta que deje de mejorar.
Pero estos cálculos pueden ser usados de otra manera. Si elegimos varios vecinos al azar y dividimos lo que mejora la solución entre la distancia del paso tendremos una manera de estimar en qué dirección las soluciones mejoran más rápido. Estamos calculando una aproximación del gradiante. En este caso podemos priorizar los que más rápido varíen.
Hay una versión de esta selección que se usaba originalmente para la metaheurística hill climbing que no requiere cálculos. Si tenemos una solución con múltiples coordenadas modificamos una a una hasta que deje de mejorar en esa coordenada. En el ejemplo anterior sería el caso de usar el vector dirección (1,0) hasta que deje de mejorar y luego usar (0,1). Tiene la desventaja de que puede quedarse atrapado cuando los mejores vecinos están en las diagonales.
Una última mejora es guardar un histórico de soluciones ya seleccionadas para evitar elegir una ya elegida o muy cercana (lo que sería el equivalente a andar en círculos). Según el coste en memoria de almacenarlas y en tiempo de compararlas puede ser mejor almacenar solo las últimas. Esta opción solo es válida cuando la selección del siguiente vecino es determinista, por ejemplo eligiendo el de mayor gradiante o cuando se inspeccionan todas las opciones y lo más probable es que se elija el mismo vecino la última vez que se «visitó» esa solución. Guardar información de los vecinos ya visitados no puede ahorrar «dar vueltas» y reducir el coste computacional.
El templado o cocido o temple o recocido simulado es una metaheurística inspirada en el templado (o cocido o temple…) de metales (en algunos sitios hablan de espadas samuráis para hacerlo parecer más interesante. Como si una metaheurística necesitara algo más para parecer interesante…mejor no respondáis). La idea es que durante el templado los átomos van colocándose en las posiciones de menor energía aunque a veces pueden moverse a una de mayor energía. Este caso depende de la temperatura a la que esté en el material y a mayor temperatura más probable es que ocurra. Tampoco todos los saltos son igual de probables, a mayor diferencia de energía también es menos probable que ocurra.
Sonará ridículo, pero uno de los problemas que he encontrado con este algoritmo es que se suelen usar términos químicos para describirlo lo cual resulta confuso para los pobres programadores que no estamos acostumbrados a ellos. Así que dejando de lado el modelo real en que se inspira, el algoritmo es similar a la búsqueda aleatoria con el añadido de que tiene cierta probabilidad de aceptar una solución cuyo valor sea peor que la actual. Es decir, que las soluciones peores que la actual no se descartan siempre, se deja al azar si se conserva la actual (la mayoría de las veces) o se remplaza por la nueva solución. Esto le permite escapar de óptimos locales explorando zonas que de otra forma no visitaría. Eso sí, para evitar ir saltando de un lado a otro a lo loco y lograr converger en un máximo la diferencia entre los valores de la función fitness no puede superar un límite que se reduce con cada iteración con lo que es menos probable que se elija una solución peor que la actual. La formula que se aplica es que una solución de fitness f’ peor que f se acepta siempre y cuando:
e ^ (f-f’/ Ti ) > rnd(0,1)
Podemos ver que se usa e ^ x para calcular la posibilidad de que se elija esa solución. Para ello calculamos la diferencia entre ambas soluciones y la dividimos entre la temperatura actual. Hay que tener en cuente que este caso solo sirve cuando busquemos el mínimo (f’ es peor si f’ > f) pero si buscamos el máximo debería de ser f’-f ( f’ es peor si f’ < f). con esto debería de darnos siempre un resultado negativo ya que si f’ es mejor que f se elegiría directamente y no llegaríamos a este punto.
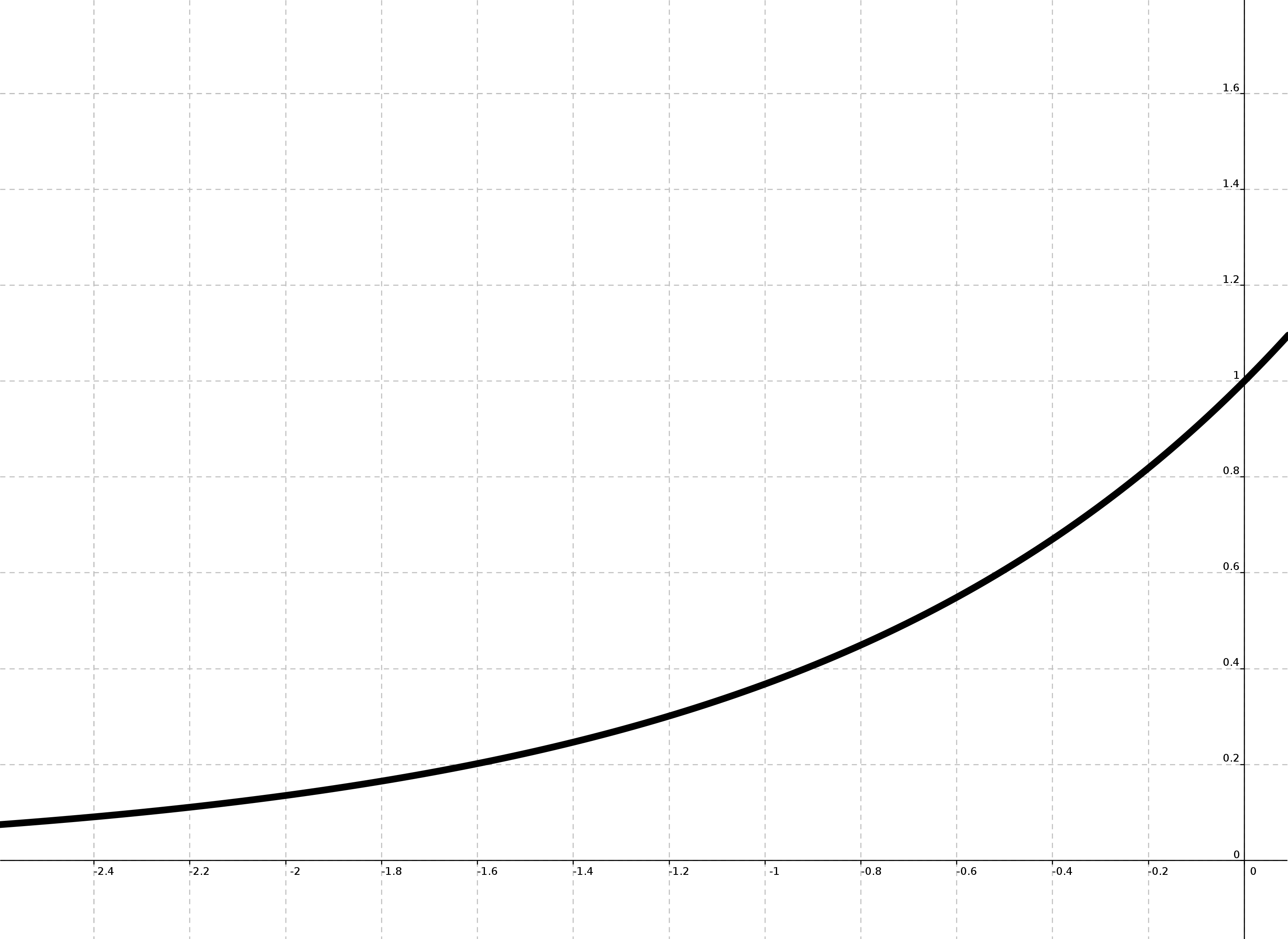
Si nos fijamos en la gráfica de la función e ^ x entre -5 y 0 vemos que su valor decrece según aumenta la diferencia entre ambos valores. También podemos ver, que al estar dividida por la temperatura, cuanto mayor sea la misma más «tolerante» es con la diferencia. Como según se itera el algoritmo la temperatura se reduce cada vez es menos probable elegir una solución peor que la actual. Según disminuye la temperatura y el comportamiento del algoritmo se asemeja más a una búsqueda aleatoria.
Gráfica de e^x para x < 0
Función de enfriamiento
La función de enfriamiento es la operación que se ocupa de reducir el valor de la temperatura cada iteración. Es más importante de lo que parece ya que es la temperatura la que permite saltar a soluciones peores y explorar el espacio de soluciones. Las dos más habituales son:
Restar un valor que sea muy pequeño «casi cero»
Ti = To – k * i = Ti = Ti-1 – k
Tiene la ventaja de que se puede calcular muy rápidamente. El problema es que es demasiado lineal y no suele dar tan buen resultado.
Multiplicar por un valor menor que uno pero cercano a él. O sea que sea «casi 1».
Ti = T0 * k ^ i = Ti = Ti-1 * k
Es rápida de calcular y da buen resultado. Variando el valor de k variamos «el comportamiento» alargando o acortando el rápido descenso inicial o lo «exploradora» que sea la metaheurística.
Algunas más complejas:
Ti = T0*k*i
Ti = (To/imax) * i
Ti = To/(1+k*ln(1+i))
La temperatura controla el umbral de aceptación de una variable. Si es alta es más fácil que una solución peor «pase el filtro». Así que lo ideal sería poder adaptarla según lo necesite el algoritmo. Acelerar su descenso, retrasarlo e incluso invertirlo. Para que el algoritmo converja en una solución es necesario que la tendencia general de la temperatura sea descender pero puede sufrir momentos puntuales de aumento. En concreto lo ideal sería que la temperatura aumente cuando esté intentando «escapar» de un óptimo local. ¿Como sabemos cuándo es eso? Sencillo, ocurre cada vez que se elige una solución peor que la actual. Nos podemos imaginar que la solución
Siendo
fbest la solución seleccionada hasta ese momento
f el valor de fitness para la solución elegida
Ti la temperatura calculada para la iteración i por alguno de los métodos anteriores
T’ = (1+(f-fbest/f))*Ti
Como f solo puede ser igual o mayor (es peor) a fbest. La formula actúa compensando la disminución de la temperatura cuando la solución empeora. De tal forma que combina ciclos de descenso con otros de ascenso aunque en teoría tendencia general es que la temperatura disminuya. ¡Ojo! Que es en teoría. Puesto que al subir la temperatura aumenta la probabilidad de que seleccione soluciones peores y cada vez que selecciona una soluciones peor aumenta la temperatura puede llegar realimentarse hasta que el aumento de temperatura se dispara.
Al final yo he optado por:
T’ = (1+(f-fbest/f))*Kinc + Ti
Al ser una suma se frena el aumento de temperatura y el parámetro Kinc permite controlar esta variación.
Aunque con los parámetros iniciales adecuados puede encontrar una correcta aproximación al óptimo global su desempeño en espacios de búsqueda muy grandes o con muchas dimensiones no suele ser muy bueno. También se usa como optimizador local.
Esta metaheurística tiene la ventaja de que es sencilla de entender y de aplicar. En su forma más simple consiste en partir de un punto aleatorio del espacio de soluciones, elegir aleatoriamente una solución cercana, si es peor se elige otra, si es mejor movernos a ella y volver a elegir otra solución desde ahí.
La implementación es fácil y su ejecución es rápida o al menos hasta donde lo permita el cálculo del fitness de la solución elegida.
var newCords = this.newSolution(this.cords.slice(0));
var newFitness = this.model.calculateFitness(newCords);
if(newFitness > this.fitness){
this.cords = newCords;
this.fitness = newFitness;
}
Por desgracia su desempeño solo es bueno en espacios de búsqueda sencillos. Es fácil que se quede atrapada en un óptimo local. Todo depende del tamaño del área en que se busca la nueva solución. Un tamaño muy grande hace que le cueste mucho más converger, uno muy pequeño causa que se quede atrapada en máximos locales. Y no hay una buena solución para todos los problemas, los valores dependen del espacio de búsqueda. Ahora es cuando estaréis pensando: «¿Y si hacemos el tamaño del área de selección variable?». Mala suerte, llegáis unas cuantas décadas tarde. Hay varios algoritmos que usan esta táctica.
Primero un detalle, el «Tamaño del área de selección» se le llama también «tamaño del paso» ya que sería el símil a lo que puede avanzar en cada iteración por el espacio de búsqueda (generalmente hay tendencia a representar el espacio de búsqueda como montañas y a nuestro algoritmo caminando por ahí).
Volviendo a las búsquedas aleatorias con «paso de tamaño variable». Hay varias estrategias, unas intentan calcular el tamaño del paso analizando la función a optimizar, otros van ajustando el valor de forma dinámica. La forma más sencilla de hacerlo es simplemente ir aumentando el tamaño del paso cada vez que compruebas un vecino y es peor que la solución actual. Una vez encuentras una solución mejor vuelves al tamaño de paso inicial. Con cuidado de no superar un tamaño máximo de paso para evitarnos problemas como superar el espacio de búsqueda.
Aunque se puede usar como optimizador global suele tener mejor desempeño como optimizador local que parte de los resultados obtenidos por un optimizador global.
Hay algunas modificaciones interesantes que mejoran algunos aspectos.
Por ejemplo, al ser el punto de origen aleatorio solo explora una pequeña parte del espacio de búsqueda. La forma más fácil de resolverlo es ejecutarlo varias veces con puntos de inicio diferentes y quedarse con la mejor. Esto permite su uso como optimizador global. Además lo hace fácilmente paralelizable. A este algoritmo se le denomina «con reinicio».
El código completo esta en este repositorio de github.